
组织驻留巨噬细胞(TRMs)是一类定居于黏膜和实质器官的免疫细胞。TRMs在维持组织稳态、促进组织生长及重塑中的关键作用已被广泛报导,然而,其在炎症早期如何因功能失调导致组织损伤,进而加速自身免疫性疾病进展的机制仍有待阐明。

2025年11月21日,浙江大学王晓健、林文龙及浙江树人大学姜敏敏共同通讯在Advanced Science在线发表题为“Tissue-Resident Macrophage-Derived E3 Ligase SMURF2 Restricts Autoimmune Inflammation by Mediating the Degradation of p-TBK1”的研究论文。该工作首次揭示了本研究揭示了SMURF2通过诱导p-TBK1泛素化降解来抑制TRM的异常增殖,调控组织稳态,抑制自身免疫性炎症。
研究者首先观察到,IBD患者及小鼠结肠组织的巨噬细胞中,E3泛素连接酶SMURF2的表达显著降低。在髓系细胞或TRM中特异性敲除Smurf2的小鼠中构建DSS诱导的结肠炎及EAE模型,相比于野生型小鼠,敲除小鼠表现出TRM过度增殖,以及更为严重的自身免疫性炎症和病理损伤。机制研究揭示,肠道屏障受损导致细菌入侵,进而通过其释放的病原相关分子模式(PAMPs)诱导了TRM中SMURF2的表达下调。进一步研究表明,SMURF2并不影响经典的LPS诱导炎症通路,而是通过与磷酸化TBK1(p-TBK1)相互作用,介导其发生Lys-27位连接的泛素化,进而经溶酶体途径促进其降解。SMURF2的缺失导致p-TBK1累积,增强了M-CSF信号通路下游(AKT/NF-κB/MAPK)的激活,从而驱动TRM异常增殖。使用TBK1特异性抑制剂Amlexanox可显著抑制TRM增殖,并逆转由SMURF2缺失引起的炎症加重。临床数据分析进一步证实,SMURF2的低表达与IBD及多种自身免疫性疾病的疾病进展密切相关。综上所述,本研究首次阐明了TRM中的SMURF2介导p-TBK1泛素化降解抑制TRM异常增殖,进而调控自身免疫性炎症,为自身免疫性疾病的治疗提供了潜力的干预靶点。
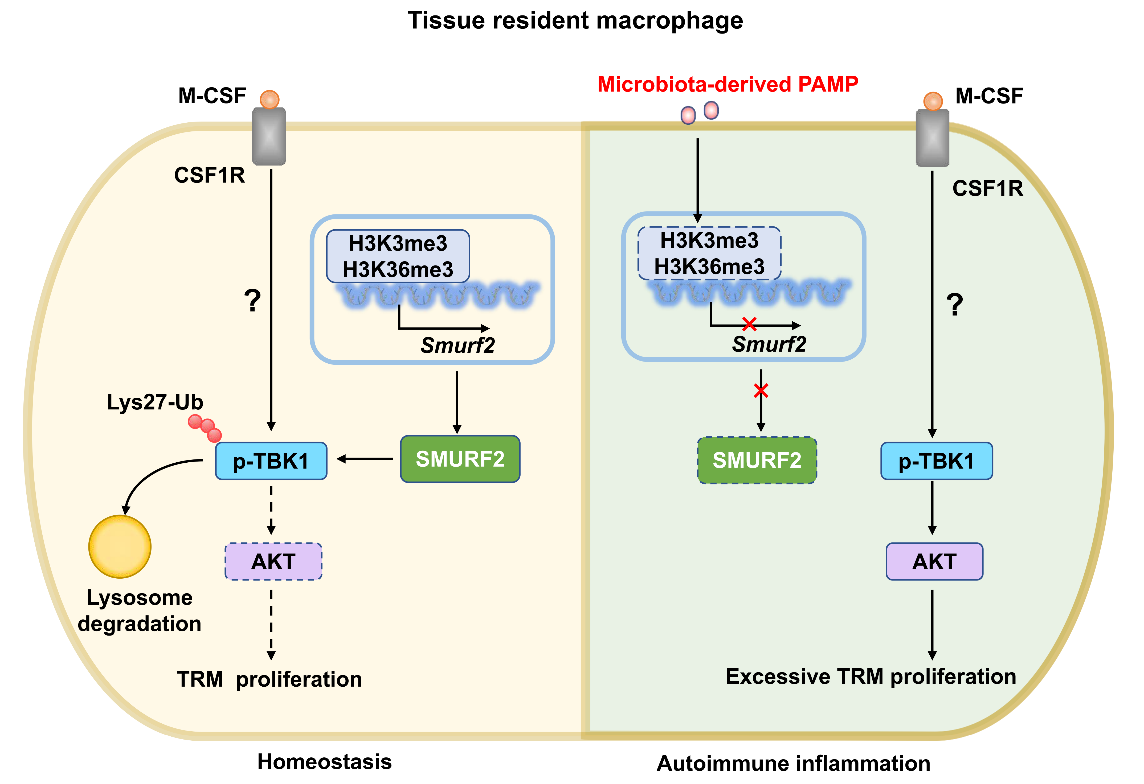
TRM细胞中SMURF2调控自身免疫病的作用模式图
浙江大学基础医学院王晓健教授,林文龙教授及浙江树人学院姜敏敏教授为本文通讯作者,课题组博士后安翔、浙一医院主任医师李君为本文共同第一作者。该文章的发表受到了国家自然科学基金的支持。课题组感谢浙江大学医学院公共技术平台的技术支持。

