
肺静脉闭塞病(PVOD)是一种发展迅速且致命的遗传性肺血管病,以肺小静脉重构和巨噬细胞中含铁血黄素沉积为主要特征,患者自确诊起生存期通常不足两年,且缺乏有效的药物治疗。目前已知大多数家族性及部分散发性PVOD病例存在EIF2AK4基因双等位突变,导致GCN2蛋白缺失,但其具体致病机制尚未明确。近日,浙江大学基础医学院杨隽教授团队在Nature Communications发表了题为Macrophage ferroptosis potentiates GCN2 deficiency induced pulmonary venous arterialization的研究论文,首次揭示了巨噬细胞铁代谢和静脉动脉化在PVOD发病中的重要作用,并为临床诊治提供了潜在新策略。 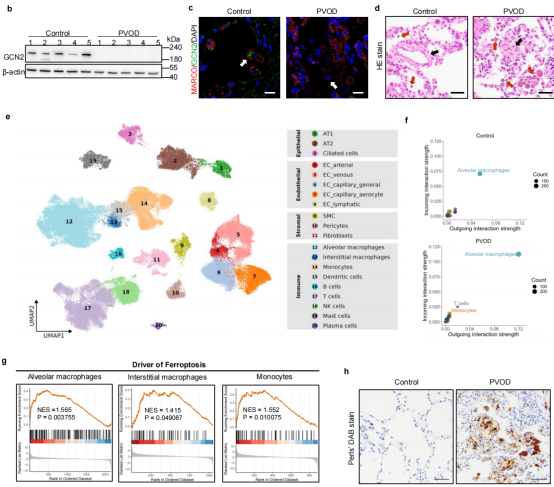
研究团队通过免疫组化和单细胞RNA测序,发现PVOD患者肺组织巨噬细胞中含铁血黄素沉积现象明显。为模拟携突变患者发病状态,团队建立了Eif2ak4K1488X/K1488X小鼠低氧PVOD模型。实验表明,铁死亡抑制剂Ferrostatin-1(Fer-1)可有效预防和逆转小鼠及MMC诱导的大鼠PVOD模型中血流动力学异常及病理改变。
通过多组学分析,团队发现PVOD患者及模型动物的肺静脉内皮细胞(VECs)中动脉特化相关基因NRP1、KDR和EFNB2表达增强,提示静脉内皮动脉化现象。为明确VEC动脉化的成因,研究者用单细胞测序发现的巨噬细胞中表达升高的GDF15、CCL3、TNF等细胞因子,及铁分别处理GCN2敲除的HUVEC,结果发现铁刺激可显著诱导动脉化基因表达上升。
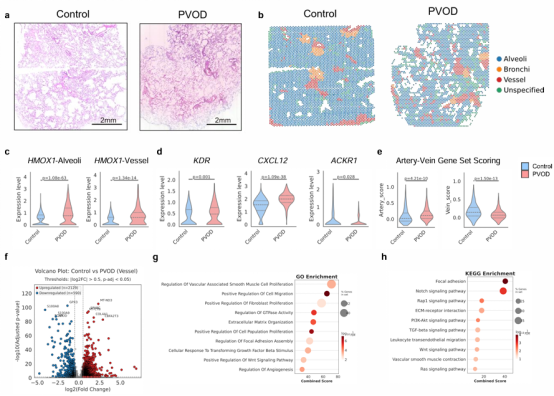
空间转录组进一步揭示,巨噬细胞铁死亡与肺静脉内皮动脉化在空间上密切相关,ETS1被鉴定为驱动NRP1/KDR/EFNB2表达的核心转录因子。机制上,组织高铁刺激ETS1表达升高,促进VECs动脉化,并伴随ERK信号通路激活。免疫荧光和功能实验均验证了p-ERK水平与动脉标志物的同步上调。此外,患者来源的iPSC分化获得的VEC亦显示出GCN2缺失背景下向动脉细胞分化的倾向,进一步佐证了上述分子机制。
综上所述,本研究系统阐明了GCN2缺失通过上调HMOX1诱导肺巨噬细胞铁死亡,继而铁释放驱动ETS1激活ERK通路,介导NRP1/KDR/EFNB2表达升高,促进静脉内皮动脉化并推动PVOD进展,可作为诊断标志。铁死亡抑制剂Fer-1可有效阻断或逆转该病理过程。这一发现不仅丰富了对PVOD发病机制的理解,也为临床治疗提供了切实可行的新思路。
该研究发表于Nature Communications,由浙江大学基础医学院生理学系、附属第二医院心血管内科、经血管植入器械全国重点实验室杨隽教授担任通讯作者,附属第二医院肺移植科陈静瑜教授和王福俤教授共同指导完成,张静圆、毛佩、周腾飞为共同第一作者,研究得到国家重点研发计划、国家自然科学基金以及医学分子生物学国家重点实验室开放课题等项目资助

