
鉴定和注释人类蛋白的序列与功能,是现代生物医学研究的重要基础。长期以来,人类基因组被认为编码约20,000种蛋白质。然而,随着核糖体印迹测序等技术的发展,越来越多证据表明,在这一经典蛋白组之外,细胞还翻译了另一个“非经典”蛋白组。它们主要由5′UTR、lncRNA等过去被视为“非编码”的RNA区段翻译而来,种类达上万种,其中许多产物长度较短,被统称为微蛋白(microprotein)。
但微蛋白研究面临一个核心悖论:一方面,许多微蛋白显示出明确的核糖体翻译信号,其中上千种微蛋白的翻译效率与经典蛋白接近;另一方面,绝大多数微蛋白在细胞内又难以被常规蛋白质组学或免疫印迹检测到。因此,解释微蛋白“活跃翻译”与 “低丰度检测”之间的矛盾,理解其翻译后的命运,是微蛋白生物学中的关键问题。
5月19日,基础医学院张汕研究员团队与生命科学学院王勇研究员团队合作,在Molecular Cell发表题为 “Intrinsic Bias of the Genetic Code Shapes the Folding and Stability Landscapes of Microproteins”的研究论文。该研究发现,大多数人类微蛋白在结构上高度无序,并在翻译后被快速降解,进一步地研究揭示了一个写在遗传密码中的组成法则:高 GC 编码序列通过遗传密码的内在偏倚富集特定氨基酸残基,从而同时塑造微蛋白的低折叠倾向、快速降解以及免疫呈递的命运。

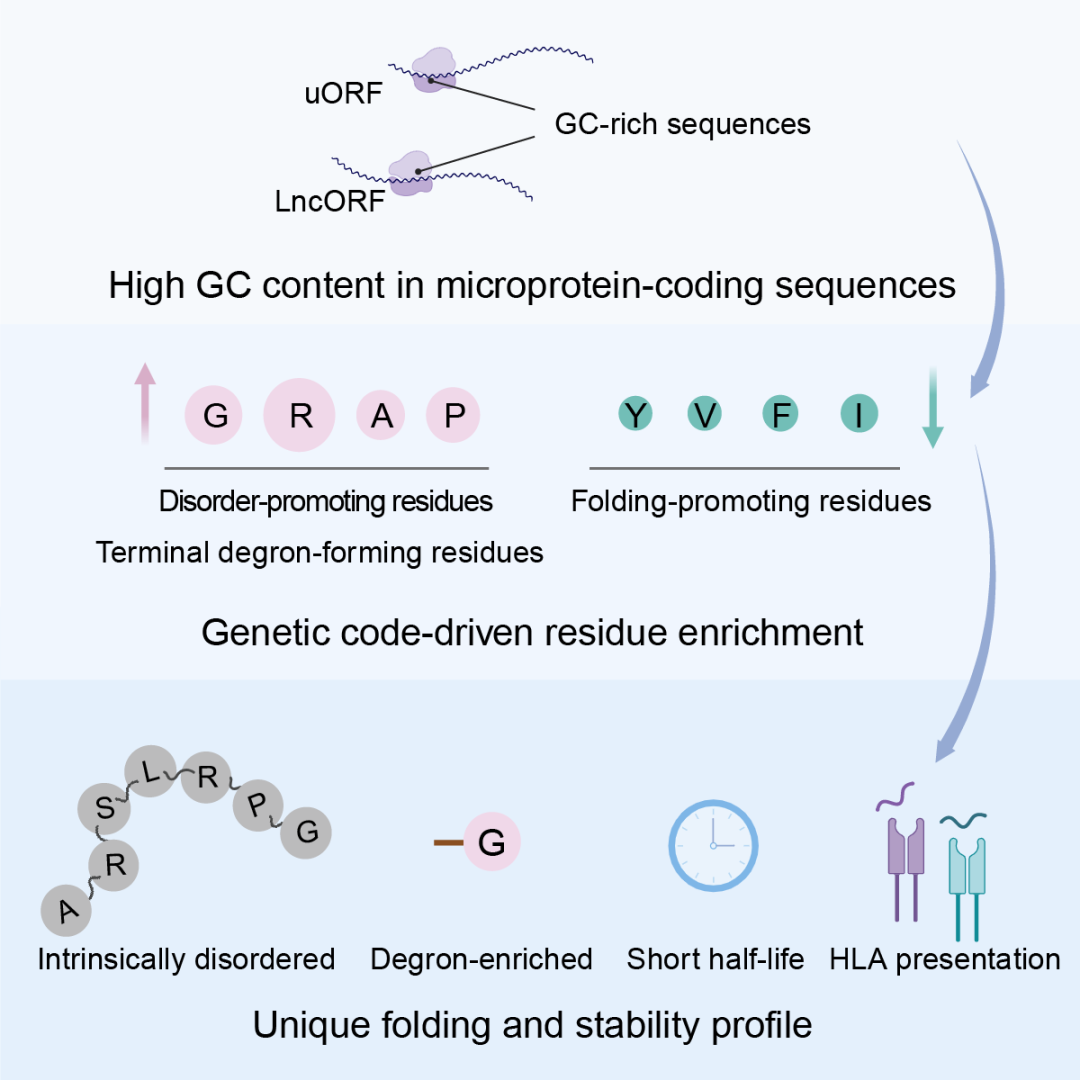
图1 微蛋白折叠与稳定性特征的决定机制
氨基酸组成塑造微蛋白折叠属性
研究团队首先整合了覆盖20余种细胞和组织的高质量核糖体图谱数据,构建了近万种人类微蛋白的集合。随后,团队利用ESMFold和AlphaFold2对这些序列进行结构预测。结果显示,绝大多数微蛋白缺乏稳定的三维折叠结构。这一结论也得到多种无序性预测工具的独立支持。
更有意思的是,微蛋白的高无序性并不是随机短序列的普遍特征。与从基因组随机区域翻译得到的序列相比,真实被翻译的微蛋白反而表现出更强的无序倾向。进一步分析表明,微蛋白富集脯氨酸(P)、甘氨酸(G)等不利于稳定折叠的氨基酸,同时相对缺乏多种疏水性残基。将微蛋白序列随机打乱、但保持氨基酸组成不变后,其无序性仍基本保留,说明整体氨基酸组成在微蛋白低折叠倾向中发挥关键作用。
微蛋白为何成为“暗物质”:蛋白质质量控制的快速清除
为了系统解析微蛋白在细胞内的稳定性,研究团队构建了多个编码上千种活跃翻译微蛋白的超长寡核苷酸文库,并将微蛋白与荧光报告蛋白通过自切割肽T2A连接。由于T2A可使二者按1:1 的化学计量比共同产生,研究者得以通过比较微蛋白信号与荧光蛋白信号,定量评估微蛋白在翻译后的稳定性。多种报告系统一致显示,大多数微蛋白在合成后迅速被降解;蛋白酶体抑制剂能够显著提高微蛋白信号,而抑制溶酶体通路影响较小,提示蛋白酶体系统是清除微蛋白的重要途径。
进一步的全基因组CRISPR筛选和文库级突变实验揭示,微蛋白序列中普遍存在两类降解信号:一类来自内部无序区域暴露的疏水残基,另一类位于蛋白质N端或C端,被称为末端降解子(terminal degron)1。例如,C端最后一位的甘氨酸(G)和丙氨酸(A),以及C端倒数第三位的精氨酸(R),均可促进微蛋白被 Cullin-RING E3泛素连接酶复合体识别并降解。通过添加C端保护序列或替换关键疏水残基,研究者能够提高大部分文库序列的稳定性,说明这些信号共同参与了微蛋白降解。
一个重要发现是,这些触发降解的末端残基在经典蛋白中受到负向选择,较少出现在暴露末端;而在微蛋白中,这种规避机制并不显著。换言之,经典蛋白在演化过程中往往已经规避了容易被质量控制系统识别的序列特征,而大量新近产生的非经典翻译产物则尚未跨越这一筛选门槛,因此更容易被蛋白质降解机器捕获。
写在遗传密码中的组成法则
既然氨基酸组成如此关键,那么微蛋白的组成偏好从何而来?研究发现,微蛋白的氨基酸组成在很大程度上(R=0.93)可以由其编码序列的GC含量解释。这一现象源于遗传密码本身的组成特征:不同氨基酸对应的密码子数量和碱基组成并不相同,其中甘氨酸(G)、精氨酸(R)、丙氨酸(A)和脯氨酸(P)由多个 GC 含量较高的密码子编码,因此会随着编码序列 GC 含量升高而显著富集。研究团队将这类残基概括为“GRAP”残基。
微蛋白编码序列之所以普遍具有较高GC含量,与其来源密切相关。许多活跃翻译微蛋白来自5′UTR,而 5′TR 本身通常富含GC;同时,高GC的RNA序列形成的复杂二级结构可放缓核糖体扫描,促进非经典翻译起始。
值得一提的是,早在979 年,研究者就注意到,低GC含量的密码子,尤其是第二位为U的密码子,偏好编码疏水性氨基酸,从而影响蛋白质折叠和质量控制。而这项研究则揭示了GC含量另一端的规则:细胞通过靶向高GC密码子编码的末端降解信号,实现对高GC序列编码的低疏水性翻译产物的清除。
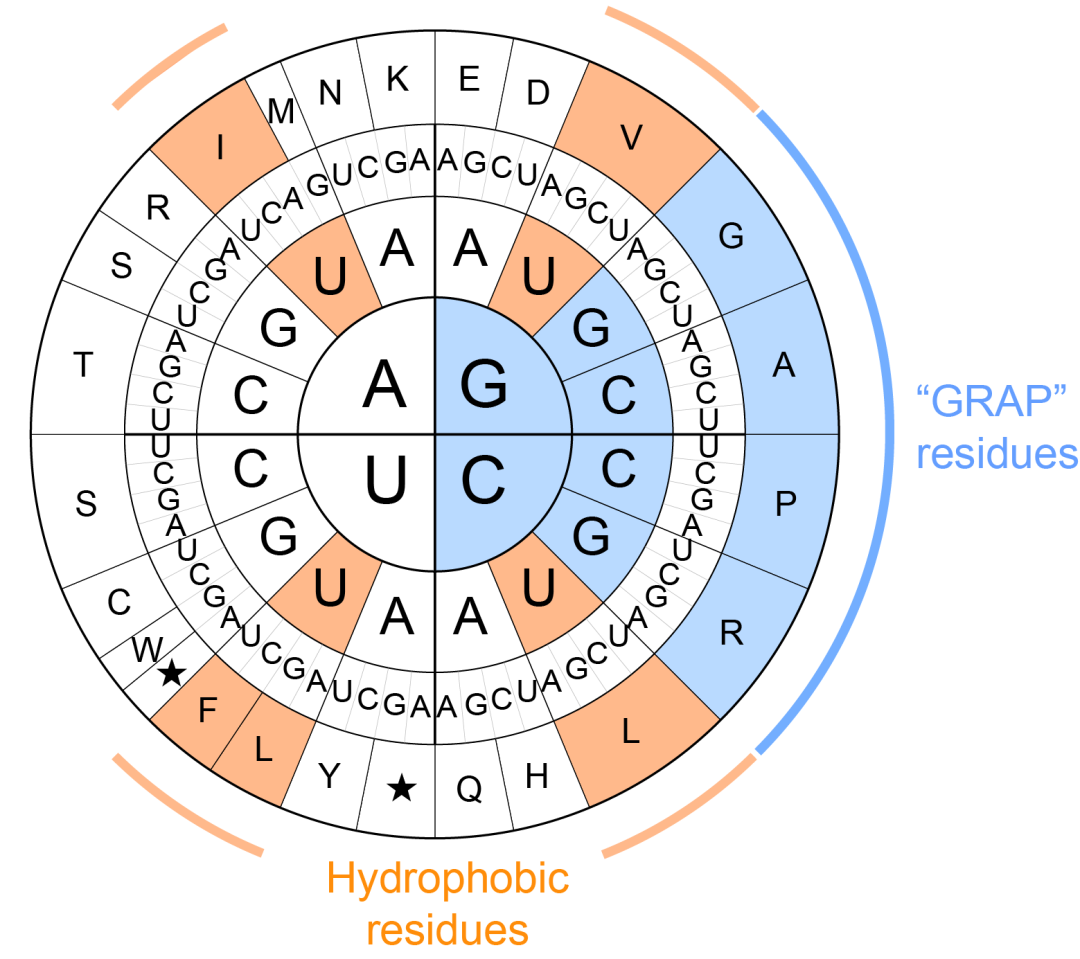
图2 遗传密码排列影响氨基酸编码偏好
降解与免疫呈递之间的内在联系
蛋白酶体降解不仅是细胞清除异常或不稳定蛋白的重要方式,也是产生HLA-I呈递肽段的重要来源。HLA-I,即人类主要组织相容性复合体I类分子,可以将细胞内蛋白降解产生的短肽展示在细胞表面,使免疫系统能够“看见”细胞内部正在发生的分子事件。因此,HLA-I呈递对于病毒感染、肿瘤抗原识别以及免疫监视都具有重要意义。最新研究表明,大量微蛋白肽段被呈递在细胞表面,占人类不同组织免疫肽组的2%–15%,并呈现出潜在的免疫调节功能。
那么,微蛋白的快速降解是否与其免疫呈递直接相关?研究团队分析了多个肿瘤类型的免疫肽组数据,发现被HLA-I呈递的微蛋白更富集C端降解信号。稳定性预测模型也显示,不稳定微蛋白更容易进入HLA-I呈递通路。此外,实验证实,与常规蛋白酶切质谱检测到的微蛋白相比,出现在HLA-I免疫肽组中的微蛋白整体稳定性更低。这些结果为微蛋白“细胞内难以检测、细胞表面却广泛呈递”的现象提供了机制解释。
综上,该研究揭示了一个连接遗传密码、蛋白折叠和蛋白质量控制的简单规则,表明编码序列核苷酸组成可以直接影响蛋白质命运,为理解功能蛋白的从头演化以及肿瘤免疫肽组来源提供了新的视角。
该研究由浙江大学基础医学院张汕研究员和浙江大学生命科学学院王勇研究员担任共同通讯作者;浙江大学基础医学院博士生郭亚波、秦啼和浙江大学生命科学学院硕士生罗健诚为共同第一作者。浙江大学医学院附属第一医院心血管内科郭晓纲教授、西安交通大学生命科学与技术学院李昊教授和华东师范大学生命科学学院曹雄文教授为本项目的数据分析提供了大力支持。本研究得到了多项国家自然科学基金的资助。

